
Research & Development
R&D POWERED BY STRUCTURE- GUIDED DRUG DESIGN
Targeting the disease at the active site
We pursue challenging diseases and design molecules with expert chemistry knowledge and multidisciplinary collaboration to help change how hereditary angioedema (HAE) and other rare diseases progress—and how a patient feels.
We are world leaders in structure-guided drug design. This specialized development approach is fundamental to how we identify and make first-in-class or best-in-class therapies that are highly potent, selective, and bioavailable.
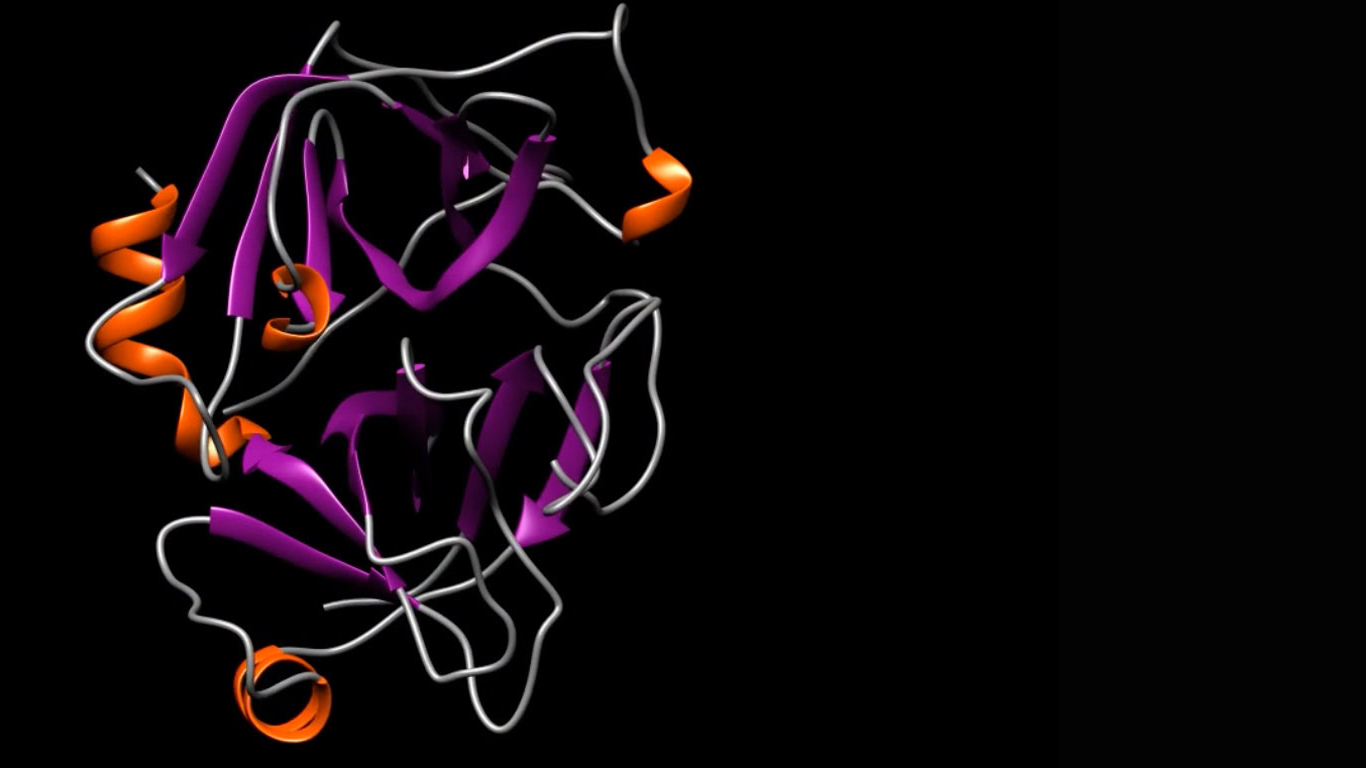
We take a multidisciplinary, stepwise approach to structure-guided drug design using an integrated application of traditional biology and medicinal chemistry
First, we leverage our experience in structural biology to select an initial target location in a protein that plays a key role in the course of a given disease. This is the target that we want to bind to—and block with—our molecule.
Then, we identify and map the three-dimensional (3D) molecular structure, such as the shape and characteristics of that active binding site.
Using that map, we then design a drug, atom by atom, that is optimized to fit that unique structure perfectly—like a hand in a glove—and suppress its biological activity.
This structure-guided drug design process requires an array of advanced technologies and computational tools, including X-ray crystallography, cryogenic electron microscopy, computer modeling of molecular structures, virtual screening, molecular docking, molecular dynamics simulations and protein chemistry to focus on the 3D structure of the active site of the target protein.
By harnessing the power of structure-guided drug design, we continue to advance a pipeline of potential best-in-class or first-in-class therapies that could make a difference in the lives of patients with complement-mediated and other rare diseases.
Areas of Focus
Hereditary Angioedema (HAE)
HAE is a potentially life-threatening rare disease caused by a genetic deficiency of a protein called C1 esterase inhibitor (C1-INH). C1-INH plays an important role in preventing the bradykinin-forming system from becoming hyperactive and mediating swelling in HAE. HAE is a rare condition, affecting between approximately 1 in 10,000 to 1 in 50,000 people.1 Left untreated, patients with HAE often have multiple attacks every month, and the swelling from each attack can last for 2 to 4 days.2
Clinical Trials
APeX-P (Active, Not Recruiting)
NCT05453968: This study is evaluating the pharmacokinetics (PK) and safety of berotralstat to determine the appropriate weight-based dose for pediatric participants 2 to < 12 years old for prophylactic treatment to prevent attacks of HAE. Learn more >>
APeX-A (Active, Enrolling by Invitation)
NCT04933721: This Phase 3b open-label study is providing access to berotralstat for patients with HAE who were previously enrolled in berotralstat studies. Learn more >>
Netherton Syndrome
Netherton syndrome is a serious, rare, lifelong genetic disorder affecting the skin, hair and immune system, caused by lack of normal function of a natural inhibitor of KLK5. People with Netherton syndrome often have red, scaly, inflamed skin, fragile hair, and are more likely to develop skin infections, allergies, asthma and eczema. Netherton syndrome can be life threatening, especially during infancy when patients are vulnerable to dehydration and recurrent infections.3 Currently, there are no approved treatments for Netherton syndrome.4
We are developing BCX17725, a potential best-in-class protein therapeutic that is designed to treat the underlying protein deficiency that causes Netherton syndrome by inhibiting KLK5, a serine protease in the skin that is unregulated in people with Netherton syndrome. Data from nonclinical studies have shown the potential for BCX17725 to achieve the potency, specificity and convenient dosing needed to treat patients with Netherton syndrome. Initial results from a Phase 1 clinical trial evaluating BCX17725 are expected in 2025.
Clinical Trials
Phase 1 study evaluating BCX17725 (Active, Recruiting)
NCT06539507: This is a first-in-human, Phase 1/1b, 4-part study that includes the evaluation of safety, tolerability, pharmacokinetics (PK), and immunogenicity of BCX17725. Learn more >>
Diabetic Macular Edema (DME)
DME is an important cause of vision loss in diabetes and is due to leakage from the blood vessels in the retina. The leakage of fluid from damaged blood vessels results in swelling and a buildup of pressure in the eye, which leads to disorganization of the retinal layers and ultimately results in vision loss.5 While current treatments focus on vascular endothelial growth factor (VEGF) inhibition, DME can develop from other mechanisms, such as the kallikrein-bradykinin pathway, a system at the cellular level that plays a vital role in human physiology. This is supported by observations that many DME patients have an incomplete response to intravitreal anti-VEGF therapies that are administered every four to eight weeks.6,7
We are developing avoralstat, a plasma kallikrein inhibitor that is designed to target the kallikrein-bradykinin system on the retinal vascular endothelial cells and may result in less vascular leakage and less edema. Using a different mechanism of action from current anti-VEGF therapies and delivered to the suprachoroidal space as a depot formulation, avoralstat is designed to provide high dose levels to the retinal vessels with long-lasting exposure. This could result in less frequent injections and a reduced burden on patients and the healthcare system.
Clinical Trials
Phase 1b study evaluating avoralstat (Active, Recruiting)
NCT07228559: This is a Phase 1b study to evaluate the safety, tolerability, and preliminary efficacy of a suprachoroidal injection of avoralstat in participants with diabetic macular edema. Learn more >>
Complement-mediated diseases
We are advancing multiple discovery programs for targets across the complement system that are approaching IND-enabling studies.
The complement system is part of the body’s natural immune system and is responsible for helping the body eliminate microbes and damaged cells. It is composed of proteins that are primarily produced in the liver and circulate in the blood. Once activated, the complement system stimulates inflammation, phagocytosis and cell lysis. Excessive or uncontrolled activation of the complement system can cause severe, and potentially fatal, immune and inflammatory disorders.
A growing pipeline of oral therapies for complement-mediated and rare diseases
BioCryst development programs represent the potential to improve the well-being of people whose lives are currently limited by complement-mediated and rare diseases. We discover novel, oral, small-molecule medicines that treat diseases in which significant unmet medical needs exist. An enzyme plays a key role in the biological pathway of the disease. Discover more >>
References
- Bernstein JA. HAE update: epidemiology and burden of disease. Allergy Asthma Proc. 2013;34(1):3-6.
- Bernstein JA. Severity of hereditary angioedema, prevalence, and diagnostic considerations. Am J Manag Care. 2018;24:S292-S298.
- Netherton syndrome. Orpha.net. Accessed August 14, 2024.
- Petrova, E., & Hovnanian, A. (2020). Advances in understanding of Netherton syndrome and therapeutic implications. Expert Opinion on Orphan Drugs, 8(11), 455–487.
- Diabetic macular oedema (DMO). MaculaSociety.org. Accessed August 14, 2024.
- Bhatwadekar, A, et al. Investigational plasma kallikrein inhibitors for the treatment of diabetic macular edema: an expert assessment. Expert Opin Investig Drugs. 2020 Mar; 29(3): 237–244.
- Diabetic Macular Edema: Diagnosis and Management. AAO.org. Accessed August 14, 2024